



Contributor: Abby Roth
Founder, Pure Microbiology
The buzz surrounding the release of USP General Chapter <797> on November 1, 2022, is undeniable, as it should be. We have seen four different possible iterations of the chapter since 2015. This highly anticipated release brings significant changes to sterile compounding operations and facility design, all with the intention of improving patient safety and ensuring patient access to essential medication. Because many of the new requirements will require extensive time to incorporate into policies and procedures, USP has set a one-year implementation date of November 1, 2023.
For those responsible for making changes to policies and standard operating procedures (SOPs), it’s time to get started. We see changes throughout the chapter, making it critical to read and reread the chapter to pick up on some very nuanced differences between the 2008 and 2022 versions. This is especially true with media-fill testing, gloved fingertip testing, and viable sampling, which will affect all sterile compounding organizations. To understand the changes to these microbiology-related tests, we must first review the newly established compounded sterile preparation (CSP) categories.
CSP Categories & BUDs
The traditional compounding risk levels of low, medium, and high have become ingrained in daily practice. But with the new chapter, they are replaced with CSP Categories 1, 2, and 3. It is important to note that low, medium, and high risk do not directly correlate with Categories 1, 2, and 3. The new categories are based on where the primary engineering control (PEC) in which a CSP is prepared, is located, and other quality-related elements, not the number or type of components used in the preparation of the sterile medication. Table 1 provides a summary of Category 1, 2, and 3 requirements. Be sure you review Section 14 of USP <797> for the full dating that can be assigned to CSPs. If you have a compounding aseptic isolator (CAI) or compounding aseptic containment isolator (CACI) in a segregated compounding area (SCA), you face a major BUD change. You are no longer afforded full dating and the CSPs you prepared are limited to the Category 1 BUDs. Because of this, many are moving away from the use of CAIs and CACIs in their SCAs and replacing them with laminar airflow workbenches (LAFWs) and biological safety cabinets (BSCs).
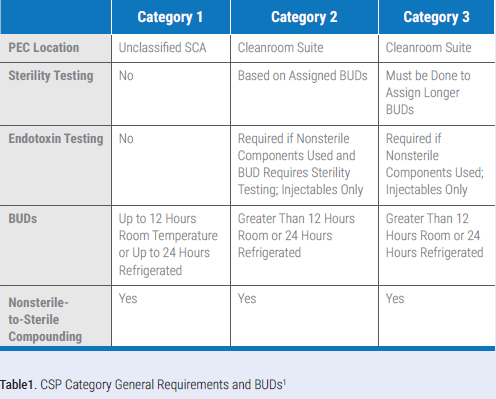
Upon review of the chapter, the longer Category 3 BUDs may seem appealing, but they come with additional requirements for:
• Personnel qualification
• Garb
• Cleaning and disinfection
• Viable sampling
• Stability determination
One other requirement, in addition to those listed, is that sterility testing must be performed on all Category 3 CSPs, supplemented with appropriate endotoxin testing. It is also important to note that Category 3 BUDs are limited to those indicated in the chapter and cannot be extended further, even with stability data.
Media-Fill & Gloved Fingertip Testing
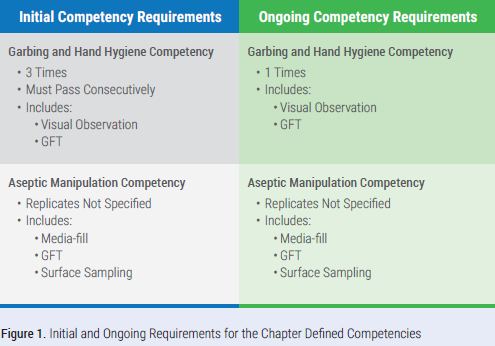
There are numerous changes related to media-fill and gloved fingertip testing (GFT) and the related competencies. The garbing and hand hygiene competency includes visual observation and GFT. Perform GFT after garbing to confirm staff can don sterile gloves without contaminating them. We also see changes to the aseptic manipulation competency where a surface sample must be collected from the work surface of the direct compounding area (DCA) after the completion of the media-fill test but before the area is cleaned and disinfected. In the event the surface sample exceeds the action level, the growth must be identified to the genus level. Identification is not required for microorganisms recovered on GFT or the media-fill test.
For the initial garbing and hand hygiene competency, it must be performed three times initially, and the staff member tested must pass consecutively. For example, if the individual passed the first two gloved fingertip tests and failed the last one, they would have to do three more tests and pass all three. For the aseptic manipulation competency, the number of replicates or total number media-fill units prepared is not defined by the chapter and the sterile compounding facility is left to decide. Figure 1 defines the required aspects of each competency.
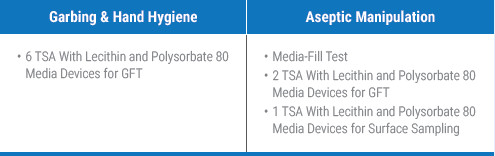
Let’s look at the numbers. For every new staff member that must complete the garbing and hand hygiene and aseptic manipulation competencies, you will minimally need 9 tryptic soy agar (TSA) with lecithin and polysorbate 80 media devices and the designated media-fill components.
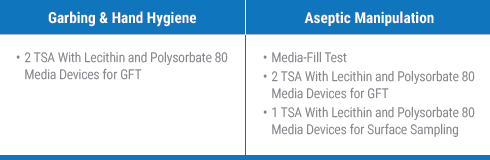
For every existing staff member that must complete the garbing and hand hygiene and aseptic manipulation competencies, you will minimally need 5 TSA with lecithin and polysorbate 80 media devices and the designated media-fill components.
One of the most impactful changes to sterile compounding operations is the increased frequency of these competencies. Annual training and competency assessments remain; however, the garbing and hand hygiene and aseptic manipulation competencies are required to be completed more frequently than what was required in the 2008 version of the chapter.3 We also see the introduction of a defined competency frequency for those who oversee compounding but do not actually compound. Table 2 defines these frequencies based on the CSP Category an individual compounds or if the individual oversees compounding.

One other major change is the incubation requirements for GFT and media-fill testing. All samples must be incubated in an incubator. GFT now has a two-stage incubation. The samples are incubated at 30 to 35 °C for at least 48 hours and then transferred to 20 to 25 °C and incubated for at least five days. Media-fill testing incubation is flexible, with the chapter providing two options as seen in Figure 2 (below).
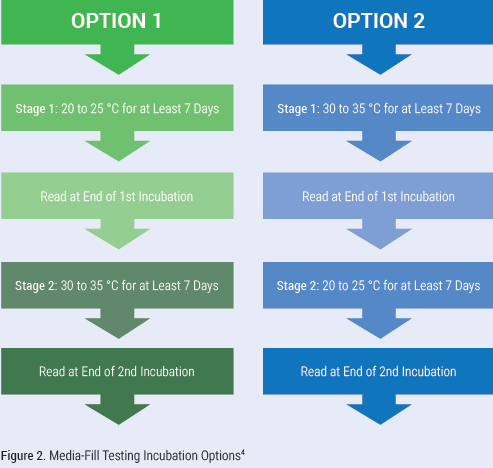
Viable Sampling
There are significant changes to viable air and surface sampling frequencies. Those compounding Category 1 and 2 CSPs are required to perform viable air sampling at least every six months and surface sampling at least monthly. Those that perform Category 3 compounding are required to collect viable air samples at least monthly and surface samples at least weekly. Table 3 defines viable sampling frequencies for all compounding categories. These increased frequencies will likely result in many sterile compounding organizations taking at least surface sampling in-house. If your organization decides to take on the collection of air or surface samples, ensure staff are trained and deemed competent in sampling technique and how to properly execute a sampling session.
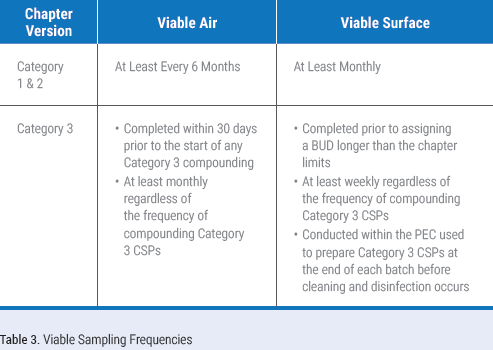
Many organizations have relied on their certification providers to choose sampling locations for them. It is critical that you evaluate your sampling plan and take responsibility for where samples will be collected. You must ensure viable air and surface samples are collected in each ISO classified space. Additionally, surface samples must be collected in the DCA of the PEC and in the pass-throughs. The chapter recommends that surface samples also be collected on equipment located in the PECs, on work areas adjacent to the PECs, and on frequently touched surfaces. Use a risk-based approach to identify viable air and surface sample locations that will provide valuable data regarding the state of control of the compounding area.
The 2008 version of the chapter required a viable air sample to be collected in the SCA in close proximity to the PEC.3 This is no longer a requirement. However, it is a recommended best practice to collect both a viable air sample and a surface sample within the perimeter of the SCA as a baseline in the event there are exceeded action levels on the samples collected in the PEC. There are no action levels for these samples and they are for information only.
The only media required to be used for sample collection is TSA. If an organization chooses, it can also use fungal media. Surface sample collection devices must contain neutralizers, such as lecithin and polysorbate 80. After collecting a surface sample, it will no longer be acceptable to wipe the surface with sterile 70% IPA. The 2022 version of the chapter will require that the surface be cleaned and disinfected after the sample is collected. The facility’s designated EPA-registered one-step disinfectant cleaner is the best option and if the sample is collected in the PEC, sterile 70% IPA would follow the application of the EPA-registered one-step disinfectant cleaner.
Viable air and surface samples must be incubated in an incubator. Incubation depends on whether one or two samples are collected at each location. Figure 3 defines the specifications for single and dual plate incubation.

The 2008 version of the chapter did not define how to count colony forming units (CFU) recovered on dual plate samples.3 The chapter now specifies that the TSA sample and TSA/fungal sample are to be treated as separate samples, all recovered growth counted, and compared to the action level. If one of the samples exceeds the action level, both should be resampled as part of the investigation and confirmation of a return to a state of microbial control.
The chapter will no longer require the identification of every CFU recovered. Instead, only samples that exceed the action levels will be required to have each colony type identified to the genus level. Additionally, the term “highly pathogenic organisms” was removed from the chapter, eliminating the need to “immediately remedy” the recovery of coagulase positive staphylococcus, gram negative rods, yeast, and mold.
Summary
These are just some of the many changes made to media-fill testing, gloved fingertip testing, and viable sampling. It is critical to read and reread the chapter to pick up on all the changes that will affect your operation. In doing so, you will likely find that there are gaps in the chapter, leaving you with more questions than answers. Utilize other industry standards and guidance documents or reach out to industry experts for help. It is also important to have multiple media suppliers, as the sterile compounding industry’s media use is about to skyrocket. Change is here. Be ready.


